In summer 2023 I did an internship at Nobias Theraputics. They develop computational tools for drug discovery.
I worked on 5 projects in total.
I did not fully complete 3 of the projects, but they were all cool so I thought I would share what I did and a short takeaway.
I will write about the two I completed in another post.
Protein Cryptic Pocket Discovery using Topological Data Analysis #
The name is a mouthful, but it is a relatively basic concept.
Many proteins have pockets (little holes) that ligands can bind to to interract (inhibit) with the protein.
Some proteins appear to not have such pockets, but under some specific conditions, a pocket appears.
The difficulty is that these conditions are not known, and simulating molecules is extermely computationally expensive. (takes days to get nanoseconds of computational molecular dynamics data)
If we could find a way to identify if a protein has a cryptic pocket, some proteins that were previously thought undruggable could possible be drugged.
Approach:
There have been many attempts to use molecular dynamics to simulate proteins, but that is extremely hard for many reasons.
My first attempt was to modify openfold to output a distribution of protein shapes from an aminoacid sequence. The thought was that if I get multiple possibilities from openfold, the non-primary shapes could be the protein shape when the cryptic pocket is visible (since a protein with a cryptic pocket has multliplle ground states).
After installing openfold and running a prediction, I found it did not output a distrbution of protin shapes so I was stuck.
I took a step back and thought about training a machine learning model to classify “has cryptic pockets” vs “no cryptic pockets”.
To do that, a model needs to take in 3D data about proteins. Nobias had developed a tool called TDA (Topological Data Analysis, specifically persistant homology) that did just that. The short summary is that it takes a point cloud of atoms and measures what kind of holes the point cloud has at different distance scales. The output looks like a barcode and encodes information about the overaall structure of the point cloud. If you want a more detailed explanation, I found a fantastic youtube video here
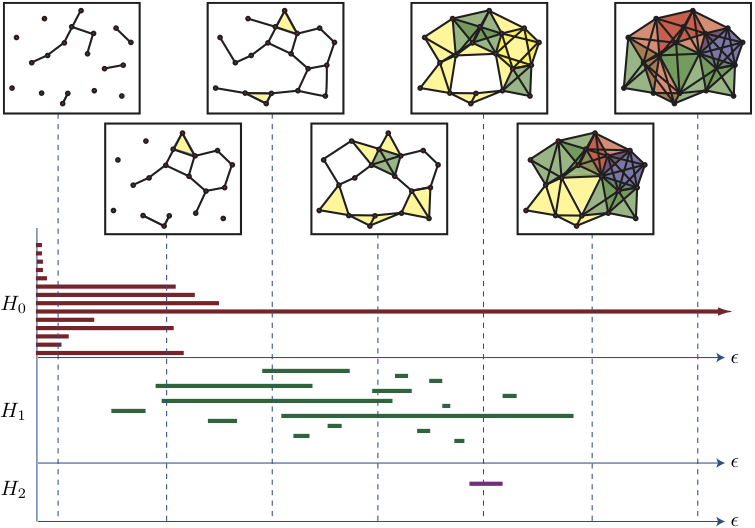
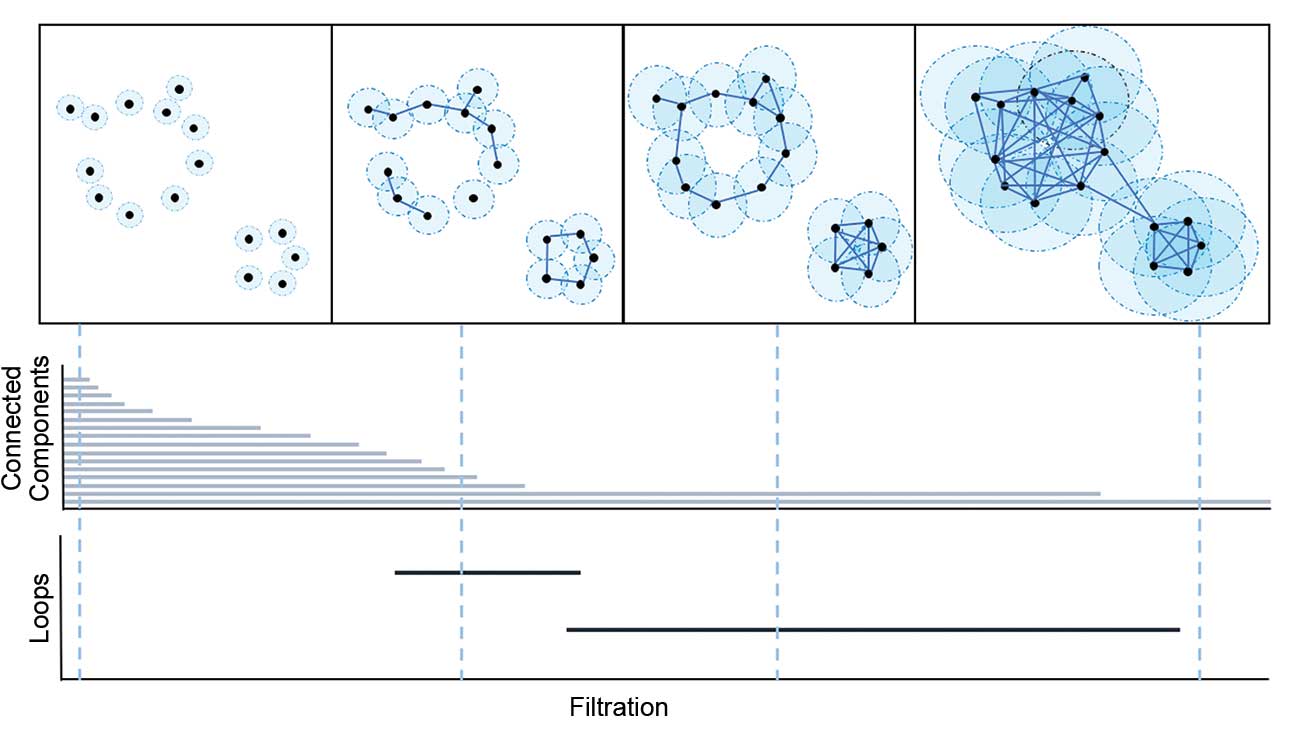
So I thought feeding this into a Machine Learning model might be able to predict a presence of a cryptic pocket.
However, since we only knew of around 300 proteins with cryptic pockets, and we did not have data on proteins without cryptic pockets, I thought there simply was not enough data.
At this point I passed the project to Andy Pheng, who was able to solve the problem with a transformer with 70% accuracy!
It turns out there was enough data to do a decent job of predicting cryptic pockets.
He dealt with the lack of data as follows:
- use 40 of the 300 examples as a test case, and use 260 for training.
- Assume well studied proteins not in the list known to have cryptic pockets to not have cryptc pockets (The logic is if they are well studied and are not known to have a cryptic pocket, they most likey don’t).
Now he had a training dataset with 260 positives and many negative examples.
Then he put these through the Nobias in house TDA program and got the TDA fingerprint (barcode looking thing) for each protein.
Then just train a small transformer on the data. To my urprise, the transformer was extremely small but was still able to get 70% AOC.
Now, using this model we can screen proteins very effectively, then perform molecular simulation on them to confirm if a pocket exists. Then feed that data back into the model as data to further improve it.
My lesson: #
Transformers are much more capable than I thought. They can learn even with relatively little data!
Permutation testing on Copy Number Variation data to identify drug targets associated with desease #
I colaborated with John Zammit on this project.
Copy Number Variations are sections of genes that are repeated. They can have effects ranging from nothing to contributions to genetic disorders.
The aim of the project is to compare CNV (copy number vairations) of healthy individuals with deseased individuals to see which regions could contribute to desease.
We decided to focus on ADHD, and use the 1000 genome database.
The statistical test we used to test the CNVs is called a permutation test. It is a simple test that tells you how likely the two groups are to come from the same distribution. A nice visual explanation of the test is here.
Unfortunately, parsing through the genetic data from the 1000 genomes project was time consuming and I had to pause my internship for a week to go to RoboCup, so I passed my project to John Zammit. He was able to continue the project and parse the data, as well as perform the permutation tests.
My lesson: #
Parsing and cleaning up big data is extremely time consuming.
Creating an Embedding network for drug names #
During the week at RoboCup, I took on another project.
The problem was that drugs have many synonyms that mean the same thing.
For example: Levocarnitine acetyl hydrochloride, 3-Acetoxy-4-(trimethylaminio)butanoic acid, AKOS024284761 mean the same compound
The idea was to create a lookup table, but the twist was it had to be differentiable. The reason was that we eventually wanted to intigrate this into a large language model.
So essentially the challenge was to create an embedding neural network that maps synonyms to the same vector. Also, a bonus feature could be that a mystyped drug name could still be recognized.
At first I tried a liner autoencoder,since it is the conventional way to create embeddings.
I downloaded list of all synonyms, then matched up all pairs of permuations of synonyms per drug in a query and answer list.
Since the model was a linear autoencoder, each character would be mapped to a number (0 - 52) assigned to the character.
This has WAYYYY too many problems… it did not work. reasons:
- the relationships between characters are NOT linear (i.e. just because a drug name has ‘a’ in the first position and another drug name has ‘b’ in the first position doesnt mean they are synonyms. But assigning 1 to ‘a’ and 2 to ‘b’ implies ‘a’ and ‘b’ are more likley to be part of the same drug than ‘a’ and ‘z’)
- The character slots further down do not get trained. This is because almost all drug names use the first 10 character slots, but very few use the last 10. This is because the input to the autoencoder needs to be the length of the longest drug name, and that is around 140 characters long! (very few drugs have more than 130 characters). If unusaed character slots are filled with 0s, the model rarely was the chance to learn the last chharacter slots.
So I tried using an image to image approach using a U-Net.
The thought was that drug names could be converted into one hot encoding picture representations, then encoded. this way there is no linear relationship implied between characters.
So I converted all the drug names in my list to 2d one hot encodings. I use da kernal size of vocabulary_size by 3 since kernales should be able to learn patterns between characters regardless of the distance between them (i.e. ‘a’ and ‘b’ vs ‘a’ and ‘z’ should be the same difficulty to learn).
I was able to train the model but as I finished training, I came back from my week at RoboCup and Nobias wanted me to work on a more fun project.
So far I havent tested the accuracy of the model but I dont have high hopes. Now I think a transformer is the ideal architecture for this since this really is not an image to image problem. At the time I did not know how to make a transformer so I tried with all the methods I knew but I should have just used a transformer.
My lesson: #
Explore many options to solve a problem before tunnel visioning into a single approach.
Going with a transformer instead of trying an Autoencoder, then a U-Net would have saved lots of time, got better results, and got me to learn transformers.